Paper by D. Pinek 2019
The paper "Unified description of the electronic structure of M2AC nanolamellar carbides" has been published in Physical Rev B.
Here you will find the paper by Damir Pinek
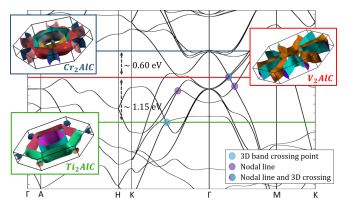
"The nanolamellar M2AC or carbon-based “211 MAX” phases, where M is an early transition metal, A belongs to groups 13–15, and C is carbon, can be described by rigid band models. The same band model applies to all possible A elements belonging to a given group of the periodic table. Changing M for a given A is then equivalent to shifting the Fermi energy EF through a band structure common to all phases in the group. This is shown by comparing predictions of density functional theory (DFT) to angle-resolved photoemission spectroscopy (ARPES) measurements. In particular, the Fermi surface of a given Al-based MAX phase can be obtained with an acceptable degree of accuracy by simply selecting the appropriate ARPES isoenergy surface of another Al-based phase. In V2AlC , and in addition to conventional metal energy bands, both DFT and ARPES show the existence of a gapped nodal line located around 0.2 eV below EF or complex crossing points at EF with Dirac-like features in some directions. Application of the rigid band model suggests that these topological features as well as others, also predicted by DFT, can be positioned at or close to EF by an appropriate choice of M and A, or by using an appropriate combination of various M and A elements."