Electronic and vibrational properties of V2C-based MXenes: from experiments to first principles modeling
Aurelie Champagne
Institute of Condensed Matter and Nanoscience (IMCN),
Abstract :
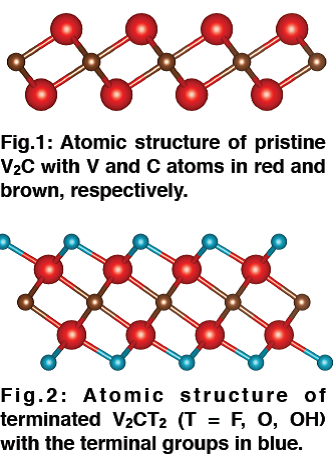
IMy presentation will focus on the characterization of V2C-based MXene systems and will deal with structural, electronic and vibrational properties. Firstly, I will describe the pristine bare V2C monolayer (see Fig.1). Using first-principles calculations, I obtain the optimized crystal structure and the relative electronic band structure. Secondly, I will show the results related to the vibrational normal modes that are calculated thanks to density functional perturbation theory (DFPT) within the harmonic approximation. I will also discuss the Raman and infrared activity of the modes and compare the former with an experimental Raman spectrum. On top of the calculations for the pristine V2C, I perform a systematic analysis of two-dimensional (2D) V2CT2 monosheets (with T = F, O, OH) (see Fig.2) with the aim to establish the role of surface terminal groups in the electronic and vibrational properties. More specifically, I will show the improvement in approximating the experimental Raman peaks positions when considering 2D fully-terminated V2CT2 systems. Finally, I will briefly present some recent results obtained for systems with mixed terminal groups. The actual agreement between our experimental and theoretical results is relatively good and some perspectives for reducing the remaining discrepancies will be proposed.
Infos date
Grenoble INP - Phelma Laboratoire LMGP 3 parvis Louis Néel - 38000 Grenoble Accès : TRAM B arrêt Cité internationale